Chan Lab:
Autophagy nutrient sensing
We are interested in understanding how a cell adapts its metabolic pathways in response to external stress. The model that we focus on is the autophagy degradative pathway in mammalian cells.
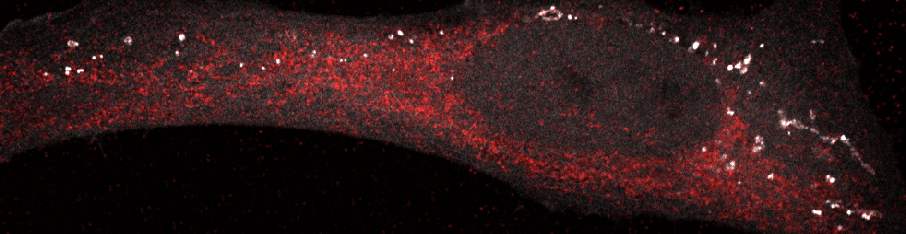
During autophagy, cellular membranes form off of the endoplasmic reticulum and elongate to capture a range of cytoplasmic targets including proteins, mitochondria or foreign bacteria. This cargo capture can be non-specific or directed by a family of ubiquitin-binding adaptor proteins that recognise components tagged for degradation. Engulfment leads to formation of a double-bilayer enclosed autophagosome, which is then transported to the lysosome. Autophagy then proceeds to a degradative phase in which metabolic building blocks are ultimately recycled back to the cell. More details are summarized in our review here.
Autophagy serves as a primary pathway for maintaining metabolic homeostasis and our work is based in 2 particular contexts that intimately depend on autophagy: normal cellular aging and cancer cell survival. Cancer cells are distinct for their re-programmed metabolic profile that features high catabolism of glucose and amino acids. Therefore, we have long been interested in the mechanisms linking amino acid and glucose sensing to autophagy.
We focus on the ATG1/ULK1 pathway for the regulation autophagy. This interest began when we identified ULK1 during siRNA screen for autophagy regulators. Since ULK1 is part of a larger gene family, further investigations involved the characterisation of ULK1/ULK2 double knockout models.
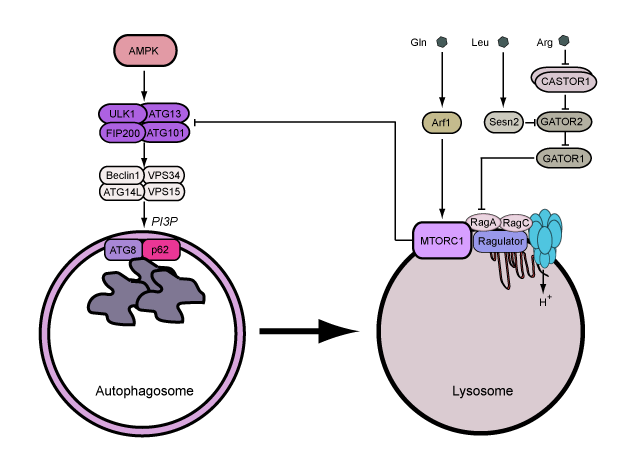
ULK1 and ULK2 are the core kinase subunits within a larger regulatory complex, which includes the subunits ATG13, ATG101 and FIP200. This ULK1/2 complex functions at the earliest stage of the autophagy signalling cascade. The ULK1 complex is interesting since it receives upstream signals from the nutrient sensor hubs MTOR complex 1 (MTORC1) and AMPK, thus combining multiple lines of metabolic stress information to autophagy regulation.
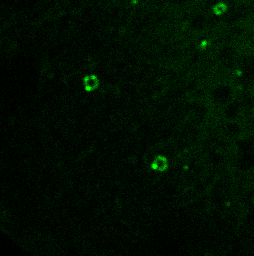
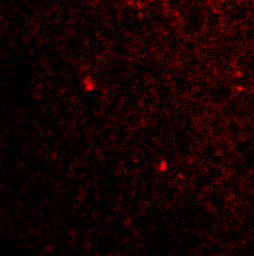
Upon integrating upstream signals, the ULK1 complex becomes activated and translocates to the endoplasmic reticulum to autophagosome assembly sites. The active ULK1 complex activates downstream autophagy signalling factors including beclin 1. Phosphorylation of beclin 1 signals activation of VPS34, which generates pools of phosphatidylinositol 3-phosphate (PI3P) at the membrane assembly site to recruit further factors that promote full autophagosome formation.
Current Projects:
Coordination of the lysosome and cell metabolism - As we studied nutrient stress regulating autophagy, we found that glucose starvation and activation of AMPK led to overall inhibition of autophagy (reported here). This novel inhibitory mechanism involved 2 separate pathways: one controlling early stages of autophagosome formation and a distinct mechanism that modulates lysosomal activation. Therefore, our work identifies a network of nutrient sensing pathways that differentially respond to amino acid and glucose. These mechanisms have multiple branches that allow the coordination of early and late autophagy stages at distinct cellular sites. We are currently investigating how amino acids and AMPK control lysosomal activity and downstream cell metabolism.
Targeting the autophagy-lysosome pathway in cancer - Autophagy generally is a pro-survival response for cells. Cancer cells, in turn, hijack this function and become ‘addicted’ to autophagy. Autophagy therefore can help cancer cells survive metabolic stress arising from high growth rates and damage caused by different treatments. An on-going strategy has been to inhibit autophagy in combination to improve the effectiveness of chemo- or radio-therapy. To date, over 60 clinical trials have been started to test the anti-malarial drug, chloroquine, to treat or sensitize drug combinations in solid and blood cancers. Since chloroquine disrupts the pH and function of the lysosome, the autophagy pathway gets blocked.
We have studied the efficiency and mechanism of chloroquine in models of triple-negative breast cancer. Contrary to the model, we found that chloroquine can kill cancer cells directly by targeting and damaging the lysosomal membrane (reported here). This work supports the idea of lysosome membrane permeabilization (LMP) as a process to promote cell death in cancer. We also unexpectedly found that glucose starvation stress alters the status of the lysosome leading to dramatic resistance to chloroquine. This pathway may cause some glucose-limited cancer cells in the core of tumours to avoid the therapeutic effects of chloroquine. We identified new modified compounds based on chloroquine that are not prone to resistance and these could be better in the clinic. We are currently aiming to further dissect the mechanism linking glucose starvation and AMPK to lysosomal function. In addition, we are developing approaches to directly target ULK1 and the autophagy pathway to inhibit cell viability in cancer.
Our recent progress in these projects have been driven by Ph.D students supported by the BBSRC, Cancer Research UK and the Nigeria Tertiary Education Trust fund.
Autophagosomes in a starved cell
PI3P lipids on the autophagosome
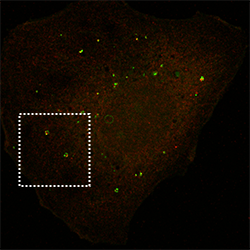
p62 autophagy cargo receptor




We thank our other funders: